Ehlers-Danlosi sündroom (EDS)
Viimati värskendatud 11 poolt Valukliinikud - Interdistsiplinaarne tervis
Ehlers-Danlosi sündroom (EDS)
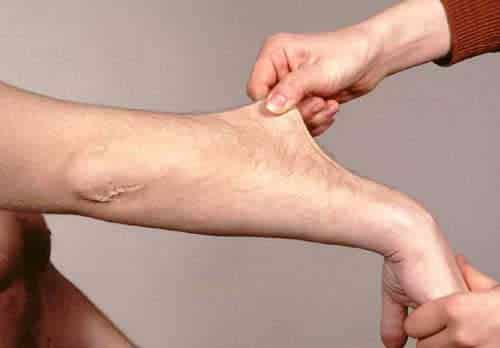
Ehlers-Danlosi sündroom on pärilik sidekoehaigus. Hinnanguliselt mõjutab Norras Ehlers-Danlos sidekoehaigust umbes 1 inimest 5000-st. Selle häire iseloomulikud sümptomid on hüpermobiilsus (ebanormaalselt elastsed ja liikuvad liigesed), hüperpainduv nahk (nahk, mis võib ulatuda tavapärasest kaugemale) ja armkoe ebanormaalne moodustumine. Seda häiret nimetatakse sageli ka hüpermobiilsuse sündroomiks (HSE). Häire sai nime kahe arsti Edvard Ehleri ja Henri-Alexandre Danlos järgi.
Samuti juhime tähelepanu sellele, et seda sidekoe häiret on mitmeid erinevaid tüüpe - sõltuvalt sellest, millist geeni või geenimoodustist see mõjutab. Need on jaotatud 6 põhikategooriasse - kuid arvatakse, et haigusel on üle 10 erineva variandi. Neil kõigil on ühine see, et neil kõigil on kollageeni (muu hulgas kõõluste ja sidemete peamine koostisosa) tootmise ja hooldamise funktsioonihäired - mis võib põhjustada tõsiseid tüsistusi.
Mõjutatud? Liitu Facebooki grupiga «Ehlers-Danlose sündroom - Norra: uuringud ja uued leiud»Selle häire uurimistöö ja meediumite kohta uusimate värskenduste saamiseks. Samuti saavad liikmed oma kogemuste ja nõuannete vahetamise kaudu igal ajal päeva jooksul abi ja tuge saada.
Sümptomid: kuidas ära tunda Ehlers-Danlosi sündroomi?
EDS-i sümptomid varieeruvad sõltuvalt teie häiretüübist. Allpool näete EDS-i kuue kõige levinuma kategooria loendit. Kõigi variantide jaoks on ühine see, et EDS on tingitud kollageeni puudumisest, talitlushäirest ja / või kahjustatud kollageenist - seetõttu on mõjutatud kollageeni sisaldavaid struktuure, sealhulgas nahka, lihaseid ja sidekude.
Põhjus: miks teil tekib Ehlers-Danlosi sündroom (EDS)?
Selle sidekoehaiguse põhjused on geneetilised tegurid. St põhjustavad pärilikud geneetilised kaasasündinud mutatsioonid. Ehlers-Danlosi sündroomi tüüp sõltub sellest, millised geenimoodustised on muteerunud.
Variandid: Mis on Ehlers-Danlosi sündroomi erinevad tüübid?
EDS on jagatud 6 põhikategooriasse. Neid klassifitseeritakse vastavalt sellele, millised geenid ja geenitüübid on muteerunud. Märgime, et paljudel erinevat tüüpi sidekoe haigustel on kattuvad sümptomid ja kliinilised nähud.
1. ja 2. tüüp (klassikaline tüüp): Sellel variandil on sageli palju samu sümptomeid ja märke kui hüpermobiilsuse rühmal (3. tüüp), kuid naha kaasatus ja sümptomid on suuremad. Mõnikord võib nende kahe rühma eristamine olla keeruline. Mutatsiooni tõttu geenides COL5A1, COL5A2, COL1A1. Mõjutab umbes 1 inimest 20000 XNUMX-st.
Tüüp 3 (hüperliikuvuse variant): Ehlers-Danlose sündroomi üks tuntumaid variante, kus hüpermobiilsuse sümptomid on kõige silmapaistvamad - ja kus nahanähud on haiguse väike osa. 3. tüüpi Ehlers-Danlose sündroomiga patsientidel esineb liigeste nihestusi oluliselt sagedamini (nt kui õlg liigest välja kukub) - nii traumaga kui ka ilma. Selle põhjuseks on vähenenud stabiilsus liigeste kõõlustes ja sidemetes; need, kes vastutavad haavatavates olukordades ja olukordades abi pakkumise eest.
Kuna seda tüüpi EDS-is on liigeste asendist välja saamine nii tavaline, on see sagedasem ka suurema valu esinemissageduse korral ning et liigeste kulumise muutused toimuvad tavapärasest varem (see tähendab, et noored võivad saada liigeste kulumise tingimusi Osteoartroos - mida tavaliselt täheldatakse ainult vanematel inimestel). Levinumad kohad, mida artroos võib mõjutada, on puusad, õlad ja alaselg, samuti kael (ülemine või alumine osa). Liigesed kuluvad seetõttu kiiremini, kuna läheduses olevates kõõlustes ja sidemetes puudub stabiilsus. 3. tüüpi EDS-i nimetatakse kattuvalt hüpermobiilsuse sündroomiks (HSE). 3. tüüp on põhjustatud mutatsioonist TNXB geenis ja epidemioloogiliste uuringute kohaselt mõjutab see umbes 1 inimest 10000 15000-XNUMX XNUMX inimesest.
Tüüp 4 (vaskulaarne Ehlers-Danlosi sündroom): Üks haruldasemaid ja surmavamaid EDS-i variante, mis hõlmavad nõrkusi arterites ja veenides - mis omakorda põhjustab suuremat tõsiste - potentsiaalselt surmaga lõppevate - komplikatsioonide, nagu veresoonte ja elundite rebenemine (rebimine), riski. Suurem osa haigestunutest diagnoositakse alles pärast nende surma.
Sellele variandile on iseloomulik, et mõjutatud inimesed on väikese kujuga ja neil on sageli väga õhuke, peaaegu poolläbipaistev nahk, kus veenid on selgesti näha sellistes piirkondades nagu rind, kõht ja muud kehaosad. Seda tüüpi EDS-iga inimestel tekivad verevalumid ka peaaegu mitte millegi eest ja verevalumid võivad tekkida ka ilma füüsiliste traumadeta.
Külgneva 4. tüüpi EDS-i raskusaste sõltub geenimutatsioonidest. Uuringud on näidanud, et umbes 25 protsendil seda tüüpi EDS-iga diagnoositud inimestel tekivad 20. eluaastaks märkimisväärsed tervisekomplikatsioonid - ja 40. eluaastaks soovitatakse, et enam kui 80 protsendil on olnud eluohtlikud komplikatsioonid. See tüüp mõjutab ligikaudu ühte inimest 1 200.000-st.
Tüüp 6 (kyphosis skolioos): See on Ehlers-Danlos eriti haruldane variant. Ainult 60 teatatud juhtumiuuringut on dokumenteeritud. EDS-i kyphosis skolioosivarianti iseloomustab skolioosi istmikuriigi järkjärguline areng, samuti silmavalgete (sklera) verevalumid ja tugev lihasnõrkus. Selle põhjuseks on geenimutatsioon PLOD1-s.
Tüübid 7A ja 7B (artrokalasia): Seda tüüpi EDS-i määrasid varem sündides mõlema puusa väga liikuvad liigesed ja nihestused (subluksatsioonid), kuid diagnostilisi kriteeriume on sellest ajast alates muudetud. See vorm on äärmiselt haruldane ja teatatud on ainult 30 juhtumist. Seda peetakse oluliselt raskemaks kui tüüp 3 (hüpermobiilsuse variant).
Tõsised tüsistused: kas Ehlers-Danlos võib olla ohtlik või surmav?
Jah, Ehlers-Danlos võib olla nii ohtlik kui ka surmav. Eriti 4. tüüpi EDS-i peetakse variantide surmavaks - seda seetõttu, et see mõjutab veresoonte süsteemi arteri ja veeni seinte nõrkusega, mis omakorda võib põhjustada aordi (peaarteri) pisaraid ja muid veritsusi. Muudel variantidel, mis ei mõjuta vaskulaarsüsteemi, võib keskmine eluiga olla täiesti normaalne. Muud tüsistused võivad olla liigesed paigast väljas ja artroosi varajane areng.
Diagnoos: kuidas tehakse Ehlers-Danlos diagnoosi?
Ehlers-Danlose diagnoos avastatakse anamneesi / anamneesi, kliiniliste uuringute põhjal ja see määratakse kindlaks geenitestide ja naha biopsia testi abil. Levinud valediagnoos võib olla kroonilise väsimuse sündroom, ME ja hüpohondriaas.
Ravi: kuidas Ehlers-Danlosit ravitakse?
EDS-i ei saa ravida. Antav ravi on ainult sümptomeid leevendav, funktsiooni tugevdav ja keskendub kahjustatud isikule sobivama funktsiooni ümber loomisele. Kuna neil, kellel on EDS, on sageli palju valu, otsivad nad lihaseid, kõõluseid ja liigeseid sageli füüsiliselt. Mõned levinumad kasutatavad raviviisid võivad olla:
- Nõelravi: lihasvalu ja müofascialsete piirangute vastu sümptomite leevendamiseks
- Füsioteraapia: nii treenimiseks, taastusraviks kui ka füsioteraapiaks
- Dieet: Õige toitumine võib neutraliseerida põletikku ning toita naha ja lihaste paranemist
- Massaaž ja lihased: Lihase- ja liigesevalu on EDS-iga mõjutatud inimeste jaoks oluline probleem
- Kohandatud liigese mobiliseerimine: liigese liikumine on oluline ja kohandatud ravi võib leevendada liigesevalu
- Kuuma vee bassein: basseinitreening on ideaalne neile, kellel on EDS
Kirurgiline protseduur: Ehlers-Danlos operatsioon
Haiguse seose tõttu ebastabiilsete liigeste ja liigesevaludega on sel rühmal märkimisväärselt suurem tõenäosus, et teda mõjutavad dislokatsioonid ja mida tuleb aeg-ajalt opereerida. Nt. õla ebastabiilsus. Selle sidekoehaiguse all kannatavate patsientide operatsioon nõuab pikema taastumisaja tõttu täiesti erinevaid ettevalmistusi ja operatsioonijärgseid kaalutlusi.
JÄRGMINE LEHEKÜLG: - Seda peaksite teadma FIBROMYALGI kohta

![]() - Jälgi julgelt Vondt.net lehte YouTube
- Jälgi julgelt Vondt.net lehte YouTube
![]() - Jälgi julgelt Vondt.net lehte FACEBOOK
- Jälgi julgelt Vondt.net lehte FACEBOOK


Suurepärane! (Suurepärane artikkel).